
Immune Mechanisms of Dietary Salt-Induced Hypertension and Kidney Disease(Abstract)
Fernando Elijovich, Thomas R. Kleyman, Cheryl L. Laffer, Annet Kirabo
Originally published7 Jul 2021https://doi.org/10.1161/HYPERTENSIONAHA.121.16495Hypertension. 2021;78:252–260
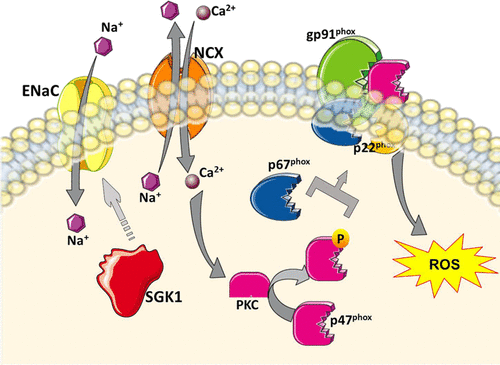
Salt sensitivity of blood pressure is an independent risk factor for cardiovascular mortality not only in hypertensive but also in normotensive adults. The diagnosis of salt sensitivity of blood pressure is not feasible in the clinic due to lack of a simple diagnostic test, making it difficult to investigate therapeutic strategies. Most research efforts to understand the mechanisms of salt sensitivity of blood pressure have focused on renal regulation of sodium. However, salt retention or plasma volume expansion is not different between salt-sensitive and salt-resistant individuals. In addition, over 70% of extracellular fluid is interstitial and, therefore, not directly controlled by renal salt and water excretion. We discuss in this review how the seminal work by Harry Goldblatt paved the way for our attempts at understanding the mechanisms that underlie immune activation by salt in hypertension. We describe our findings that sodium, entering antigen-presenting cells via an epithelial sodium channel, triggers a PKC (protein kinase C)- and SGK1 (serum/glucocorticoid kinase 1)-stimulated activation of nicotinamide adenine dinucleotide phosphate oxidase, which, in turn, enhances lipid oxidation with generation of highly reactive isolevuglandins. Isolevuglandins adduct to proteins, with the potential to generate degraded peptide neoantigens. Activated antigen-presenting cells increase production of the TH17 polarizing cytokines, IL (interleukin)-6, IL-1β, and IL-23, which leads to differentiation and proliferation of IL-17A producing T cells. Our laboratory and others have shown that this cytokine contributes to hypertension. We also discuss where this sodium activation of antigen-presenting cells may occur in vivo and describe the multiple experiments, with pharmacological antagonists and knockout mice that we used to unravel this sequence of events in rodents. Finally, we describe experiments in mononuclear cells obtained from normotensive or hypertensive volunteers, which confirm that analogous processes of salt-induced immunity take place in humans.
Hypertension research continues being of the utmost importance since hypertension is the worldwide leading cause of mortality and disability, accounting for 10.8 million or 19.2% of all attributable deaths in 2019.1 Also, control rates in the United States had reached 53.8% in 2013 to 2014, but declined to 43.7% in 2017 to 2018, for several reasons.2 Among them, 6% of all patients with hypertension have truly resistant hypertension, and 10% of the latter have the most severe pattern of refractory hypertension.3 Finally, even controlled hypertension is associated with increased residual cardiovascular risk of unclear cause.4
Harry Goldblatt, a forefather of hypertension research, was stimulated by observations as a clinician (lack of hypertension in a patient dying of uremia after accidental removal of a horseshoe kidney) and as a pathologist (presence of renal arteriolar abnormalities in the autopsies of hypertensive patients) to hypothesize that hypertension required the presence of the kidneys. To explore the controversy on whether the renal arteriolar abnormalities were the cause or consequence of hypertension, he decided to mimic the situation of renal ischemia by constricting the main renal arteries of dogs. He was very cognizant of the difference between such a model and the intrarenal vascular abnormalities of essential hypertension but proceeded with it nonetheless as the most feasible method to reproduce the suspected mechanism of renal ischemia.5
The revolutionary consequences of the results of his experiments were 2-fold. First, the recognition that renal artery stenosis was sufficient to produce sustained blood pressure (BP) elevation, later leading to the recognition of renovascular hypertension in humans. Most importantly, after preliminary investigations on possible mechanisms, Goldblatt postulated that one or more humoral factors produced by the kidneys needed to be involved. A few years later, Page and Braun Menendez simultaneously identified Ang II (angiotensin II)6,7 which began a century of additional research on the role of this peptide in BP regulation.
Gavras et al8 showed that dependency of BP on the renin-angiotensin system in rat models of Goldblatt hypertension was linked to the status of salt balance (natriuresis by the unclipped kidney with persistent hyperreninemia in the unilateral model [2K-1C], as opposed to sodium [Na+] retention and renin suppression in the bilateral one, 1K-1C).9 The group of Laragh at Cornell proposed that Goldblatt’s findings were, therefore, applicable to essential hypertension because the heterogeneous arteriolar lesions of this disorder resulted in some ischemic glomeruli with impaired Na+ excretion, and other nonischemic, hyperfiltering ones with enhanced natriuresis.10
The interplay between Ang II and salt in BP regulation acquired a new dimension once it was demonstrated that tissue-generated Ang II played ubiquitous roles in growth, proliferation and organ damage, central nervous system regulation of sympathetic tone, and most importantly, in renal Na+ transport by direct actions on transporters and by stimulation of aldosterone release.
More recently, a series of seemingly unrelated observations suggested participation of immunity and inflammation in the pathogenesis of hypertension. For example, hypertension was produced in normal rats by transfer of lymphocytes from rats with renal infarction or from splenocytes of rats with deoxycorticosterone acetate-salt hypertension.11,12 Conversely, immunosuppressants, thymectomy, antithymocyte serum, or transplant of a normal thymus prevented or reversed hypertension in several hypertensive rodent models.11,13–15 A direct link between these observations and the previously recognized interplay between Ang II and Na+ was provided by Guzik et al,16 who showed that Ang II and deoxycorticosterone acetate-salt hypertension were attenuated in mice genetically lacking lymphocytes.
Other investigators showed that Na+ is involved in immune disorders unrelated to hypertension, for example, experimental encephalomyelitis.17 We embarked on the investigation of how Na+ may trigger immune changes that underlie the pathophysiology of hypertension.